
单细胞分析雕刻师--常见整合方法比较(二)
Preface
随着单细胞测序技术的迅猛发展和市场的不断下沉,越来越多的研究人员都青睐于使用该技术来阐明一些生物学或医学问题,使通过传统bulk-RNA测序无法解决的事情得以实现成为可能,如细胞图谱的绘制、稀有细胞的鉴定与识别、细胞发育/分化轨迹的构建、肿瘤的精细化研究等。与此同时,也产生了海量的单细胞数据,而这些数据通常来源于不同的实验室,具有不同的构建时间、不同的操作人员以及不同的试剂批次等等。上述差异往往会对数据的合并造成严重的影响,导致批次效应的出现,进而干扰对真实的生物学效应的鉴别,因此,如何将不同来源的数据完美地系在一起一直是一个复杂的、具有挑战性的问题。在过去的十几年间,有数十种数据整合方法相继被开发出来,它们基于不同的原理或应用场景实现对数据的合并,在保留生物学差异的同时尽可能地去除批次效应。这里,我们选择了一些比较常见的工具或方法,包含ComBat、BBKNN、Seurat CCA、Seurat RPCA、Harmony、LIGER、fastMNN、Conos、Scanorama总共9种,通过应用于同一套数据对其进行比较。
上期《单细胞分析雕刻师--常见整合方法比较(一)》为大家带来了4类常见的整合方法,本期推送继续为大家带来Harmony、fastMNN等更多整合方法。
Results
05 Harmony
Harmony[3]使用迭代聚类方法来对齐不同数据集的细胞 (Figure7)。该算法首先将数据结合起来,并使用PCA将数据投影到一个低纬空间中,然后,Harmony使用一个迭代程序来去除批次效应。每一次迭代包括四个步骤:
1)使用一种自定义的k-means软聚类方法将细胞聚类;
2)为每个聚类计算一个全局质心,为每个特定数据集计算一个质心;
3)使用上步结果计算每一个数据集的矫正因子;
4)最后,依据细胞特定因子——一组经过加权的数据集矫正因子的线性组合——来矫正细胞。
重复步骤1~4直到收敛。该方法返回的是细胞的低纬嵌入。我们使用默认参数运行 harmony::RunHarmony ,并选取结果的前30个主成分(此处指返回的Harmony向量)进行细胞聚类及其它分析。
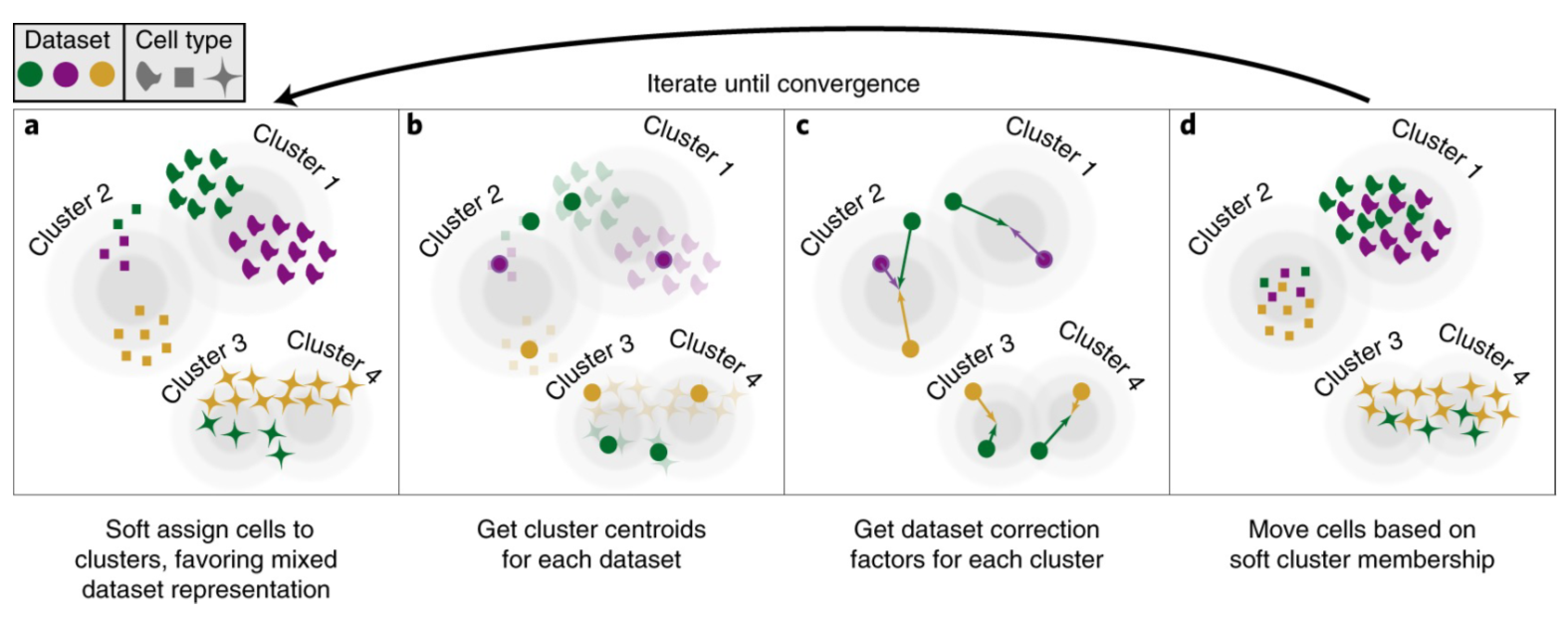
Figure7| Harmony算法模式图。使用PCA将细胞嵌入低纬空间,Harmony在该降维空间中执行迭代程序。

Figure8| Harmony 整合分析结果。左边为UMAP降维图形展示,分别以数据集和细胞类型分组;右图是结果评分。
06 fastMNN
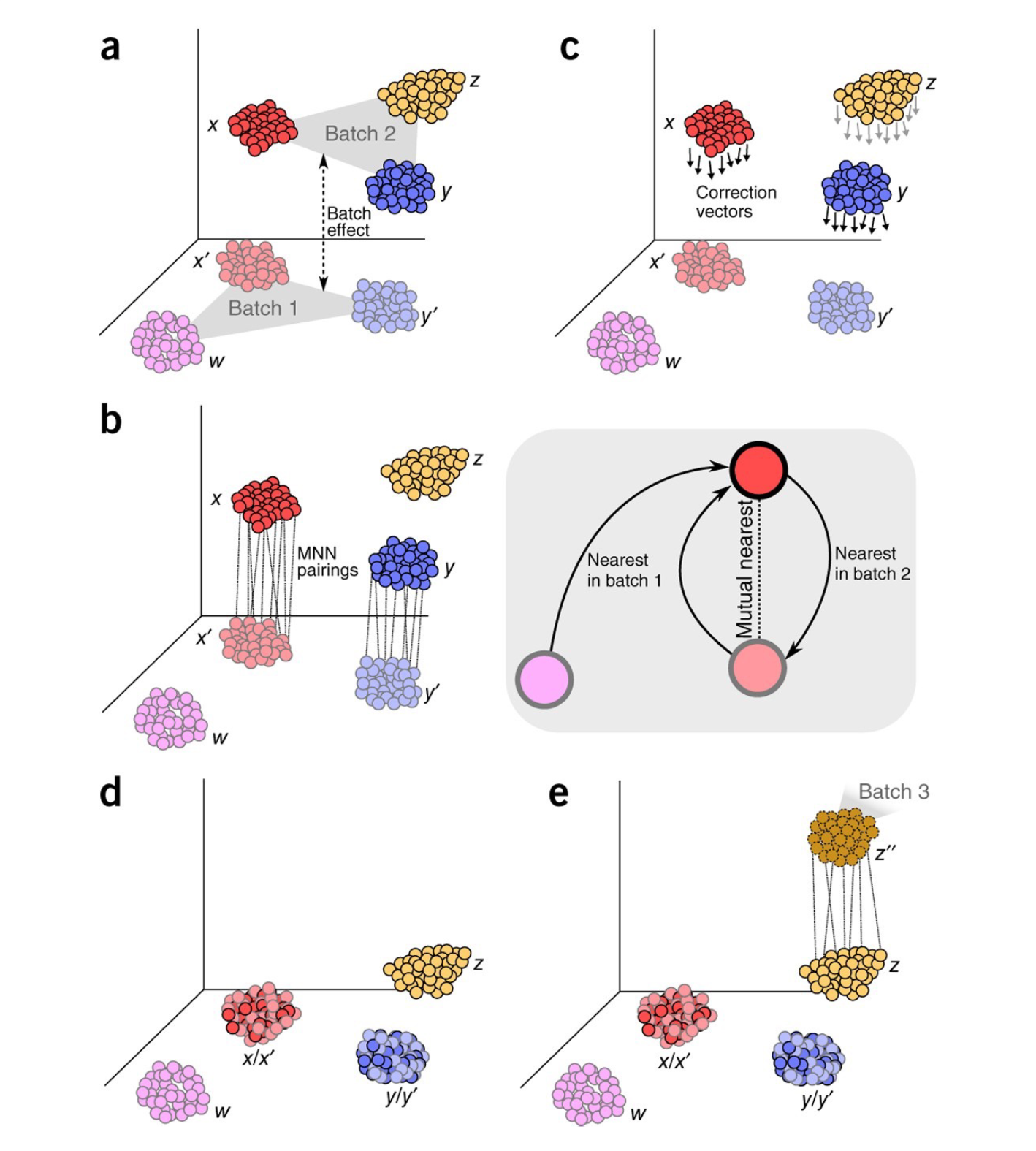
Figure9| MNN算法模式图。该算法通过寻找两个高维数据Batch1和Batch2之间的MNNs(灰色方框)确定细胞对,Batch1作为Reference,从而计算矫正向量,然后应用到Batch2中并且合并到Batch1中。整合后的数据可作为新的Reference
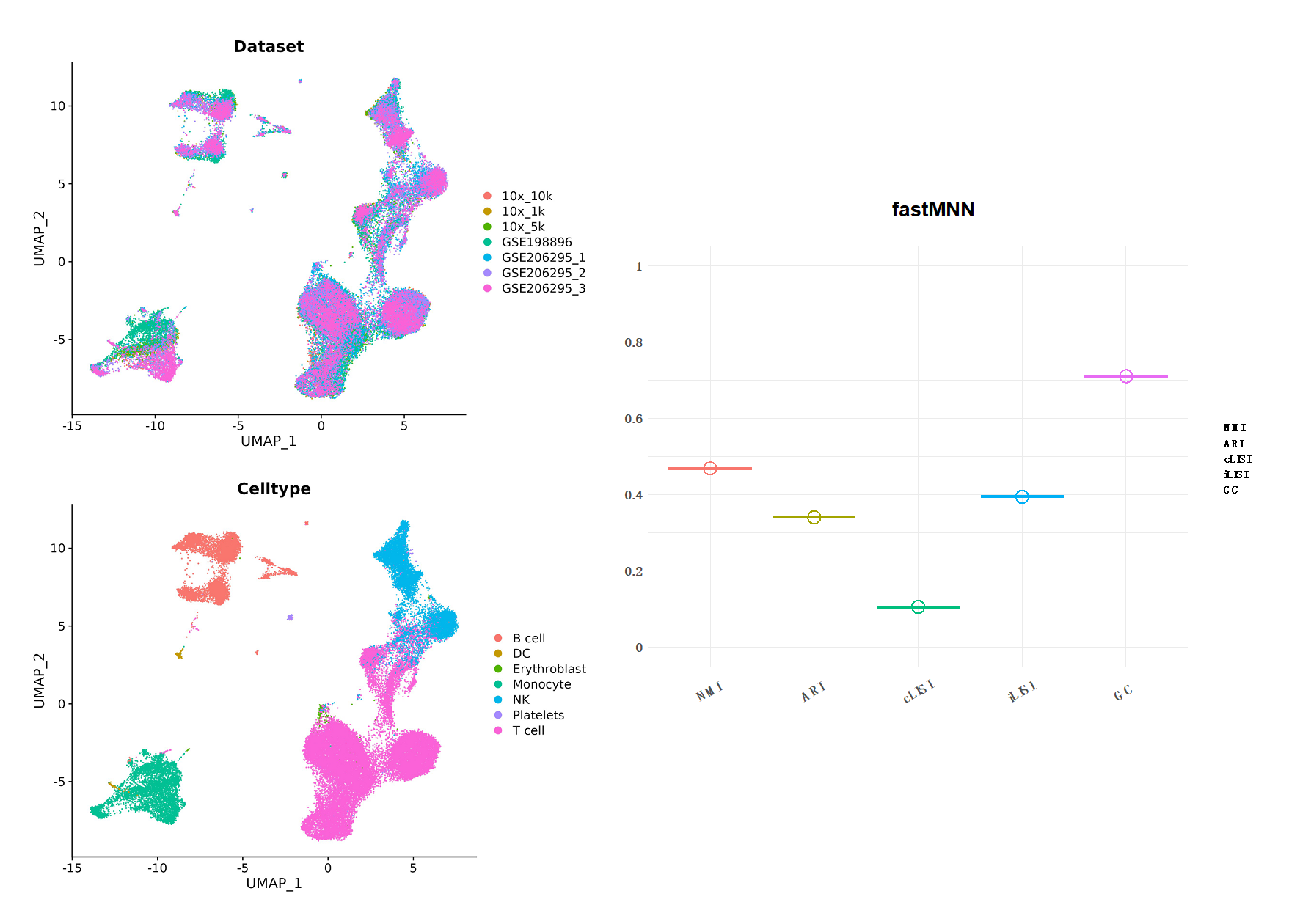
Figure10| fastMNN 整合分析结果。左边为UMAP降维图形展示,分别以数据集和细胞类型分组;右图是结果评分
07 BBKNN
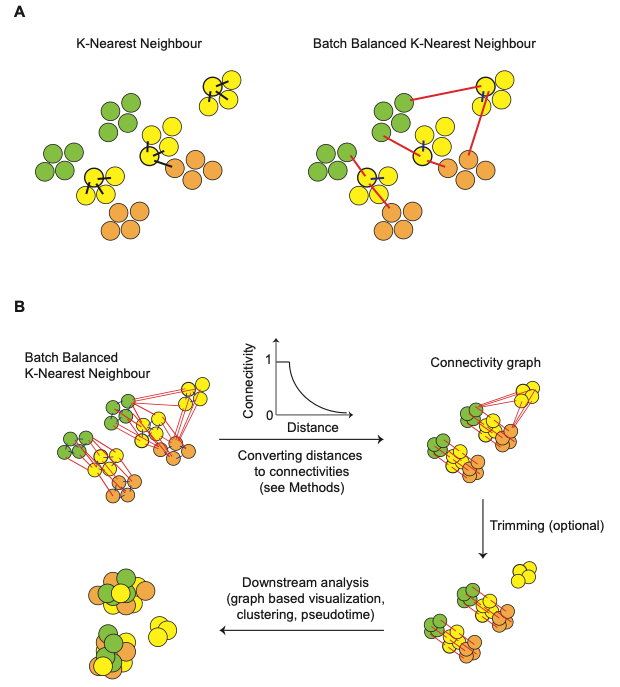
Figure11| BBKNN算法模式图。图A简单展示了一个细胞的kNN及BBKNN的结果,对每一个细胞,BBKNN会计算其在每个批次中的最近邻。图B展示了图形的构建过程,将距离转换成指数相关的连接,同时对结果进行修剪,移除错误连接
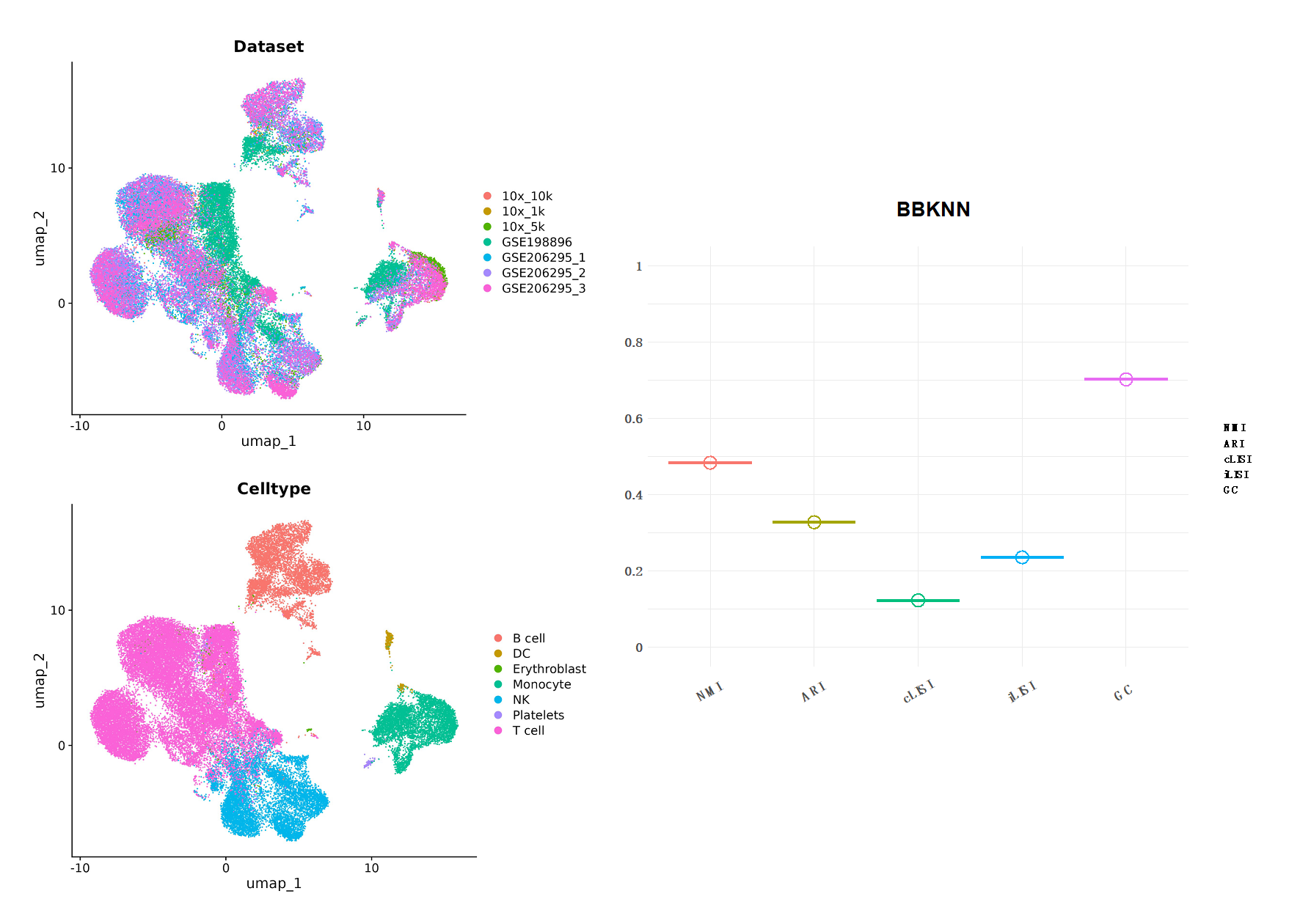
Figure12| BBKNN 整合分析结果。左边为UMAP降维图形展示,分别以数据集和细胞类型分组;右图是结果评分
08 LIGER
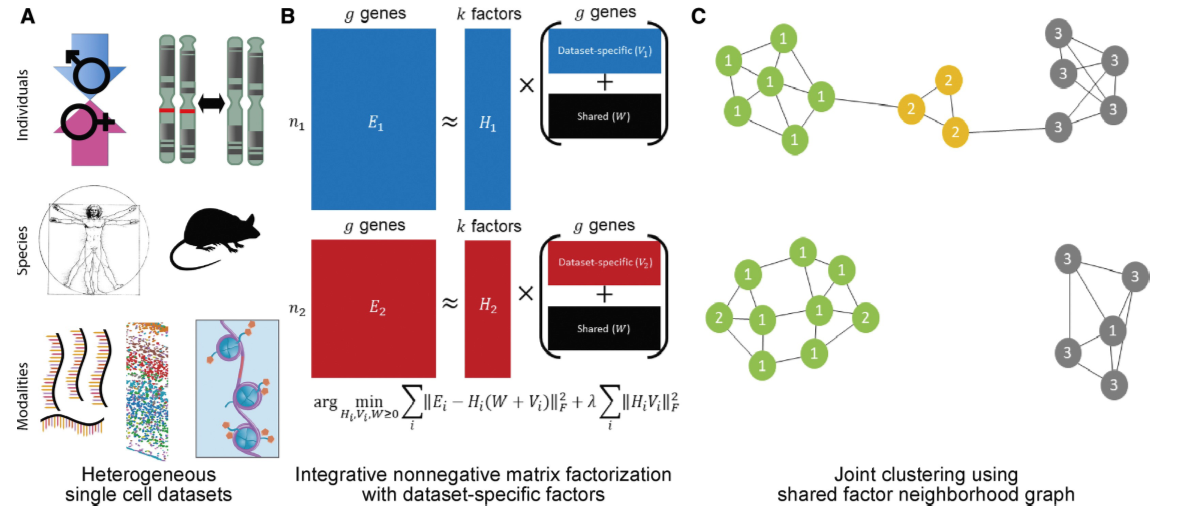
Figure13| Liger整合示意图。图A:LIGER可以以多种类型数据作为输入;图B:iNMF方法识别共享和数据集特异的基因;图C:通过iNMF得到的低纬嵌入空间进行图构建。每一个细胞都依据最大因子负载被分配上一个标签,并且连接到其最近邻上,然后通过衡量比较邻近因子负载值进行重分析以防止错误整合
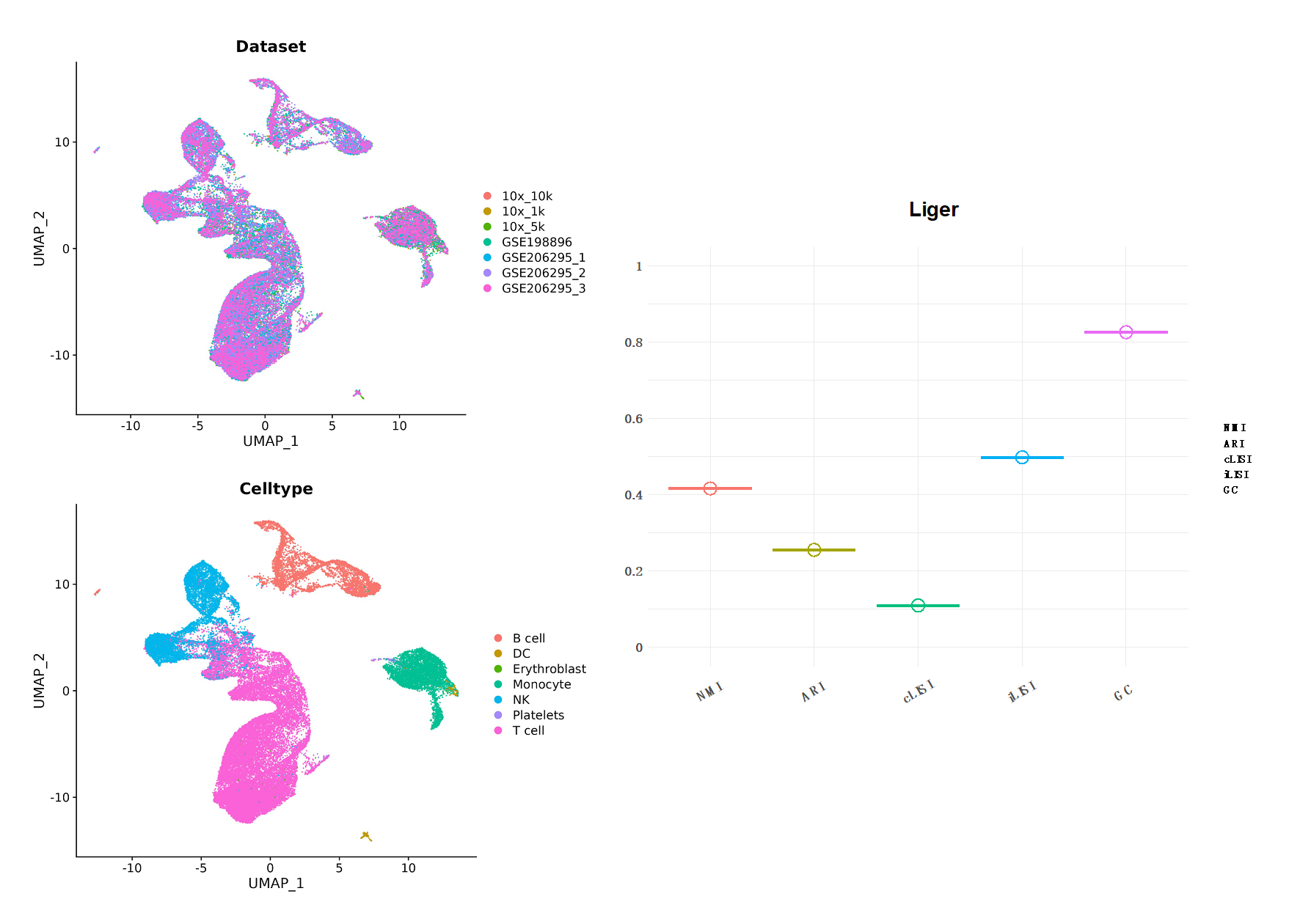
Figure14| LIGER 整合分析结果。左边为UMAP降维图形展示,分别以数据集和细胞类型分组;右图是结果评分
~ 其他整合方法请见下回分解 ~
